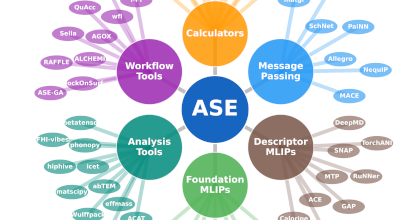
Welcome to the homepage of the ASE community. The Atomic Simulation Environment (ASE) provides a Python-based platform for setting up, manipulating, running, visualizing, and analyzing atomistic simulations. ASE is a free and open source software package developed by a large international community of scientists and research engineers. The strategic leadership and governance for the ASE project is provided by the ASE Steering Committee.