
Note
Go to the end to download the full example code.
Structure optimization: H2O#
Let’s calculate the structure of the H2O molecule. Part of this tutorial is exercise-based, so refer back to what you learnt from ASE Introduction: Nitrogen on copper and Atoms and calculators. Suggested solutions to the exercises are found after each exercise, but try solving them youself first!
Exercise
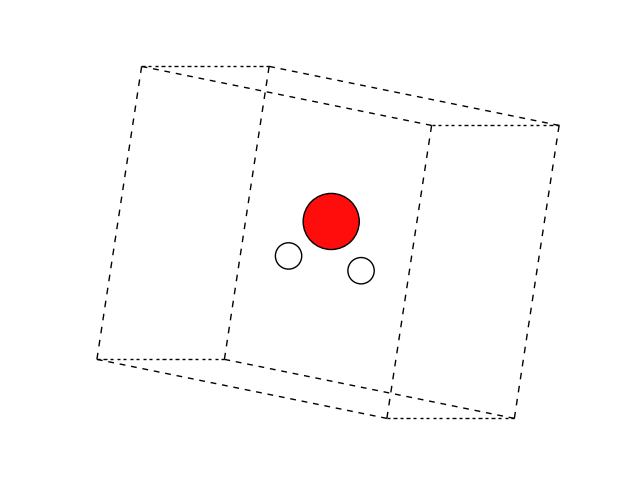
Create an Atoms object representing an H2O
molecule by providing chemical symbols and a guess for the positions.
Visualize it, making sure the molecule is V shaped.
Solution:
import matplotlib.pyplot as plt
from ase import Atoms
from ase.visualize.plot import plot_atoms
atoms = Atoms('HOH', positions=[[0, 0, -1], [0, 1, 0], [0, 0, 1]])
atoms.center(vacuum=3.0)
fig, ax = plt.subplots()
plot_atoms(atoms, ax, rotation='10x,60y,0z')
ax.set_axis_off()
Exercise
Run a self-consistent calculation of the approximate H2O molecule using GPAW.
Solution:
Optimizers#
We will next want to optimize the geometry.
ASE provides several optimization algorithms
that can run on top of Atoms equipped with a calculator:
Exercise
Run a structure optimization, thus calculating the equilibrium geometry of H2O.
Solution:
np.True_
The trajectory keyword above ensures that the trajectory of intermediate
geometries is written to opt.traj.
Exercise
Visualize the output trajectory and play it as an animation. Use the mouse to drag a box around and select the three atoms — this will display the angles between them. What is H–O–H angle of H2O?
Solution:
Note that the above will open in a separate graphical window. As always in ASE, we can do things programmatically, too, if we know the right incantations:
103.45874999690383
38.27062500154801
38.27062500154817
The documentation on the Atoms object provides
a long list of methods.
G2 molecule dataset#
ASE knows many common molecules, so we did not really need to type in
all the molecular coordinates ourselves. As luck would have it, the
ase.build.molecule() function does exactly what we need:
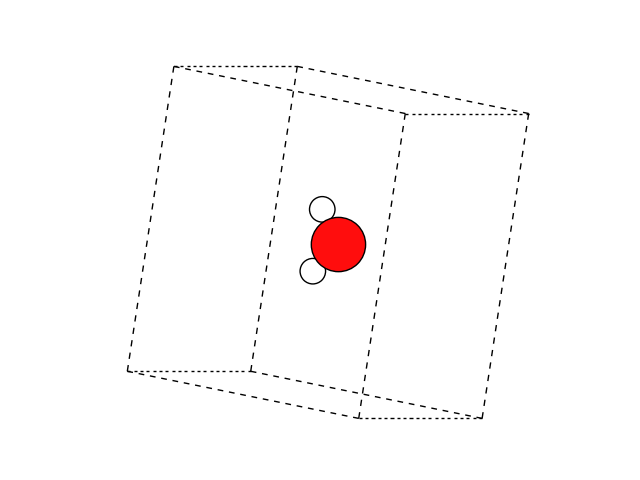
This function returns a molecule from the G2 test set, which is nice
if we remember the exact name of that molecule, in this case ‘H2O’.
In case we don’t have all the molecule names memorized, we can work
with the G2 test set using the more general ase.collections.g2
module:
['PH3', 'P2', 'CH3CHO', 'H2COH', 'CS', 'OCHCHO', 'C3H9C', 'CH3COF', 'CH3CH2OCH3', 'HCOOH', 'HCCl3', 'HOCl', 'H2', 'SH2', 'C2H2', 'C4H4NH', 'CH3SCH3', 'SiH2_s3B1d', 'CH3SH', 'CH3CO', 'CO', 'ClF3', 'SiH4', 'C2H6CHOH', 'CH2NHCH2', 'isobutene', 'HCO', 'bicyclobutane', 'LiF', 'Si', 'C2H6', 'CN', 'ClNO', 'S', 'SiF4', 'H3CNH2', 'methylenecyclopropane', 'CH3CH2OH', 'F', 'NaCl', 'CH3Cl', 'CH3SiH3', 'AlF3', 'C2H3', 'ClF', 'PF3', 'PH2', 'CH3CN', 'cyclobutene', 'CH3ONO', 'SiH3', 'C3H6_D3h', 'CO2', 'NO', 'trans-butane', 'H2CCHCl', 'LiH', 'NH2', 'CH', 'CH2OCH2', 'C6H6', 'CH3CONH2', 'cyclobutane', 'H2CCHCN', 'butadiene', 'C', 'H2CO', 'CH3COOH', 'HCF3', 'CH3S', 'CS2', 'SiH2_s1A1d', 'C4H4S', 'N2H4', 'OH', 'CH3OCH3', 'C5H5N', 'H2O', 'HCl', 'CH2_s1A1d', 'CH3CH2SH', 'CH3NO2', 'Cl', 'Be', 'BCl3', 'C4H4O', 'Al', 'CH3O', 'CH3OH', 'C3H7Cl', 'isobutane', 'Na', 'CCl4', 'CH3CH2O', 'H2CCHF', 'C3H7', 'CH3', 'O3', 'P', 'C2H4', 'NCCN', 'S2', 'AlCl3', 'SiCl4', 'SiO', 'C3H4_D2d', 'H', 'COF2', '2-butyne', 'C2H5', 'BF3', 'N2O', 'F2O', 'SO2', 'H2CCl2', 'CF3CN', 'HCN', 'C2H6NH', 'OCS', 'B', 'ClO', 'C3H8', 'HF', 'O2', 'SO', 'NH', 'C2F4', 'NF3', 'CH2_s3B1d', 'CH3CH2Cl', 'CH3COCl', 'NH3', 'C3H9N', 'CF4', 'C3H6_Cs', 'Si2H6', 'HCOOCH3', 'O', 'CCH', 'N', 'Si2', 'C2H6SO', 'C5H8', 'H2CF2', 'Li2', 'CH2SCH2', 'C2Cl4', 'C3H4_C3v', 'CH3COCH3', 'F2', 'CH4', 'SH', 'H2CCO', 'CH3CH2NH2', 'Li', 'N2', 'Cl2', 'H2O2', 'Na2', 'BeH', 'C3H4_C2v', 'NO2']
To visualize the selected molecule as well as all 162 systems, run
view(atoms)
view(g2)
Use another calculator#
We could equally well substitute
another calculator, often accessed through imports like from
ase.calculators.emt import EMT or from ase.calculators.aims import
Aims. For a list, see ase.calculators or run:
$ ase info --calculators