
Note
Go to the end to download the full example code.
Bulk Structures and Relaxations#
Here, we create bulk structures and optimize them to their ideal bulk properties
import matplotlib.pyplot as plt
import numpy as np
from ase.build import bulk
from ase.calculators.emt import EMT
from ase.eos import EquationOfState
from ase.filters import FrechetCellFilter
from ase.optimize import BFGS
from ase.units import kJ
from ase.visualize.plot import plot_atoms
Setting up bulk structures#
ASE provides three frameworks for setting up bulk structures:
ase.build.bulk(). Knows lattice types and lattice constants for elemental bulk structures and a few compounds, but with limited customization.
ase.spacegroup.crystal(). Creates atoms from typical crystallographic information such as spacegroup, lattice parameters, and basis.
ase.lattice. Creates atoms explicitly from lattice and basis.
Let’s run a simple bulk calculation. We use ase.build.bulk()
to get a primitive cell of silver, and then visualize it. Silver is known
to form an FCC structure, so presumably the function returned a primitive
FCC cell. You can, e.g., use the ASE GUI to repeat the structure and
recognize the A-B-C stacking.
atoms = bulk('Ag')
fig, ax = plt.subplots()
plot_atoms(atoms * (3, 3, 3), ax=ax)
ax.set_xlabel(r'$x (\AA)$')
ax.set_ylabel(r'$y (\AA)$')
fig.tight_layout()
# For interactive use of the ASE GUI:
# view(atoms * (3,3,3))
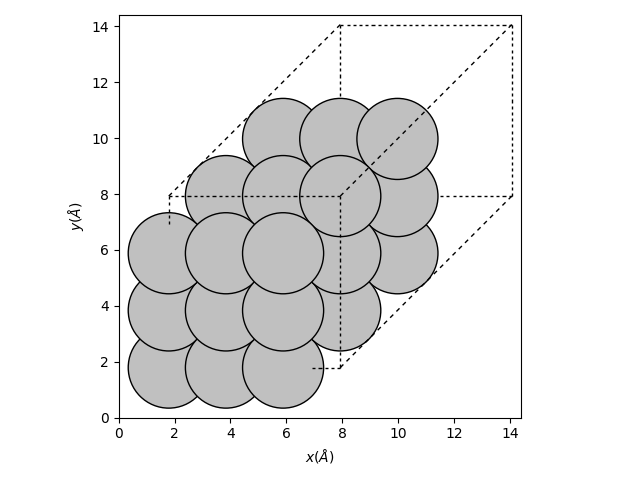
ASE should also be able to verify that it really is a primitive FCC cell and tell us what lattice constant was chosen:
print(f'Bravais lattice: {atoms.cell.get_bravais_lattice()}')
Bravais lattice: FCC(a=4.09)
Periodicity#
Periodic structures in ASE are represented using atoms.cell
and atoms.pbc.
* The cell is a Cell object which represents
the crystal lattice with three vectors.
* pbc is an array of three booleans indicating whether the system
is periodic in each direction.
print('Cell:\n', atoms.cell.round(3))
print('Periodicity: ', atoms.pbc)
Cell:
[[0. 2.045 2.045]
[2.045 0. 2.045]
[2.045 2.045 0. ]]
Periodicity: [ True True True]
Equation of state#
We can find the optimal lattice parameter and calculate the bulk modulus by doing an equation-of-state calculation. This means sampling the energy and lattice constant over a range of values to get the minimum as well as the curvature, which gives us the bulk modulus.
The online ASE docs already provide a tutorial on how to perform equation-of-state calculations: (EOS: Introduction to the Equation of state)
First, we calculate the volume and potential energy, whilst scaling the
atoms’ cell. Here, we use ase’s empirical calculator EMT:
calc = EMT()
cell = atoms.get_cell()
volumes = []
energies = []
for x in np.linspace(0.95, 1.05, 5):
atoms_copy = atoms.copy()
atoms_copy.calc = calc
atoms_copy.set_cell(cell * x, scale_atoms=True)
atoms_copy.get_potential_energy()
volumes.append(atoms_copy.get_volume())
energies.append(atoms_copy.get_potential_energy())
Then, via ase.eos.EquationOfState(), we can calculate and plot
the bulk modulus:
eos = EquationOfState(volumes, energies)
v0, e0, B = eos.fit()
ax = eos.plot()
ax.axhline(e0, linestyle='--', alpha=0.5, color='black')
ax.axvline(v0, linestyle='--', alpha=0.5, color='red')
print(f'Minimum Volume = {v0:.3f}AA^3')
print(f'Minimum Energy = {e0:.3f}eV')
print(f'Bulk modulus = {B / kJ * 1.0e24:.3f} GPa')
plt.show()
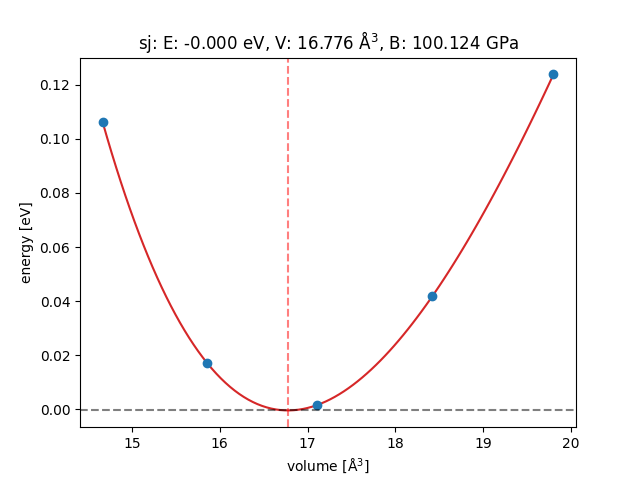
Minimum Volume = 16.776AA^3
Minimum Energy = -0.000eV
Bulk modulus = 100.124 GPa
Bulk Optimization#
We can also find the optimal, relaxed cell via variable-cell relaxation.
This requires setting a filter, in this case the
ase.filters.FrechetCellFilter() filter, which allows minimizing both the
atomic forces and the unit cell stress.
original_lattice = atoms.cell.get_bravais_lattice()
calc = EMT()
atoms.calc = calc
opt = BFGS(FrechetCellFilter(atoms), trajectory='opt.Ag.traj')
opt.run(fmax=0.05)
print('\n')
print(f'Original_Lattice: {original_lattice}')
print(f'Final Lattice: {atoms.cell.get_bravais_lattice()}')
print(f'Final Cell Volume: {atoms.get_volume():.3f}AA^3')
Step Time Energy fmax
BFGS: 0 16:01:18 0.001584 0.198735
BFGS: 1 16:01:18 0.000255 0.113074
BFGS: 2 16:01:18 -0.000366 0.003126
Original_Lattice: FCC(a=4.09)
Final Lattice: FCC(a=4.06315)
Final Cell Volume: 16.770AA^3