
Note
Go to the end to download the full example code.
Surface adsorption study using the ASE database#
In this tutorial we will adsorb C, N and O on 7 different FCC(111) surfaces with 1, 2 and 3 layers and we will use database files to store the results.
See also
The ase.db module documentation.
Bulk#
First, we calculate the equilibrium bulk FCC lattice constants for the seven
elements where the EMT potential offers a
computational affordable calculator. We store the resulting configurations
together with its bulk modulus data in the database bulk.db:
from pathlib import Path
from ase import Atoms
from ase.build import add_adsorbate, bulk, fcc111
from ase.calculators.emt import EMT
from ase.constraints import FixAtoms
from ase.db import connect
from ase.eos import calculate_eos
from ase.io import write
from ase.optimize import BFGS
bulk_syms = ['Al', 'Ni', 'Cu', 'Pd', 'Ag', 'Pt', 'Au']
path_to_bulk_db = Path('bulk.db')
# clean up if there is an old file
path_to_bulk_db.unlink(missing_ok=True)
# use context manage to ensure that connection is closed afterwards
with connect(path_to_bulk_db) as bulk_db:
for symb in bulk_syms:
atoms = bulk(symb, 'fcc')
atoms.calc = EMT()
eos = calculate_eos(atoms)
v, e, B = eos.fit() # find minimum
# do one more calculation at the minimum and write to database:
atoms.cell *= (v / atoms.get_volume()) ** (1 / 3)
atoms.get_potential_energy()
bulk_db.write(atoms, bm=B)
Here is how we can inspect the results of the previous step:
$ ase db bulk.db -c +bm # show also the bulk-modulus column
id|age|formula|calculator|energy| fmax|pbc|volume|charge| mass| bm
1|10s|Al |emt |-0.005|0.000|TTT|15.932| 0.000| 26.982|0.249
2| 9s|Ni |emt |-0.013|0.000|TTT|10.601| 0.000| 58.693|1.105
3| 9s|Cu |emt |-0.007|0.000|TTT|11.565| 0.000| 63.546|0.839
4| 9s|Pd |emt |-0.000|0.000|TTT|14.588| 0.000|106.420|1.118
5| 9s|Ag |emt |-0.000|0.000|TTT|16.775| 0.000|107.868|0.625
6| 9s|Pt |emt |-0.000|0.000|TTT|15.080| 0.000|195.084|1.736
7| 9s|Au |emt |-0.000|0.000|TTT|16.684| 0.000|196.967|1.085
Rows: 7
Keys: bm
$ ase gui bulk.db
The bulk.db is an SQLite3 database in a single file:
$ file bulk.db
bulk.db: SQLite 3.x database
If you want to see what’s inside you can convert the database file to a json file and open that in your text editor:
$ ase db bulk.db --insert-into bulk.json
Added 0 key-value pairs (0 pairs updated)
Inserted 7 rows
or, you can look at a single row like this:
$ ase db bulk.db Cu -j
{"1": {
"calculator": "emt",
"energy": -0.007036492048371201,
"forces": [[0.0, 0.0, 0.0]],
"key_value_pairs": {"bm": 0.8392875566787444},
...
...
}
The json file format is human readable, but much less efficient to work with compared to a SQLite3 file.
Adsorbates#
Now we do the adsorption calculations and store the results
in the ads.db database:
path_to_ads_db = Path('ads.db')
ads_syms = ['C', 'N', 'O']
nlayers = [1, 2, 3]
def optimize_adsorbate(symb, alat, nlayer, ads):
atoms = fcc111(symb, (1, 1, nlayer), a=alat)
add_adsorbate(atoms, ads, height=1.0, position='fcc')
# Constrain all atoms except the adsorbate:
fixed = list(range(len(atoms) - 1))
atoms.constraints = [FixAtoms(indices=fixed)]
atoms.calc = EMT()
opt = BFGS(atoms, logfile=None)
opt.run(fmax=0.01)
return atoms
# clean up if there is an old file
path_to_ads_db.unlink(missing_ok=True)
# again use context-manager
with connect(path_to_ads_db) as ads_db:
for row in bulk_db.select():
alat = row.cell[0, 1] * 2 # lattice constant
symb = row.symbols[0]
for nlayer in nlayers:
for ads in ads_syms:
atoms = optimize_adsorbate(symb, alat, nlayer, ads)
myid = ads_db.reserve(layers=nlayer, surf=symb, ads=ads)
if myid is not None:
ads_db.write(
atoms, id=myid, layers=nlayer, surf=symb, ads=ads
)
We now have a new database file with 63 rows:
$ ase db ads.db -n
63 rows
These 63 calculations only take a few seconds with EMT. Suppose you want to use DFT and send the calculations to a supercomputer. In that case you may want to run several calculations in different jobs on the computer. In addition, some of the jobs could time out and not finish. It’s a good idea to modify the script a bit for this scenario. Therefore we added a couple of lines to the inner loop.
The reserve() method will check if there is a row
with the keys layers=n, surf=symb and ads=ads. If there is, then
the calculation will be skipped. If there is not, then an empty row with
those keys-values will be written and the calculation will start. When done,
the real row will be written into to the reserved row with id=myid.
This modified script can run in several jobs all running in parallel and no
calculation will be done twice.
In case a calculation crashes, you will see empty rows left in the database:
$ ase db ads.db natoms=0 -c ++
id|age|user |formula|pbc|charge| mass|ads|layers|surf
17|31s|jensj| |FFF| 0.000|0.000| N| 1| Cu
Rows: 1
Keys: ads, layers, surf
Delete them, fix the problem and run the script again:
$ ase db ads.db natoms=0 --delete
Delete 1 row? (yes/No): yes
Deleted 1 row
$ python ads.py # or sbatch ...
$ ase db ads.db natoms=0
Rows: 0
Reference energies#
Let’s also calculate the energy of the clean surfaces and the isolated
adsorbates and store them in the refs.db database:
path_to_refs_db = Path('refs.db')
def clean_surface_reference(symb, alat, nlayer):
atoms = fcc111(symb, (1, 1, nlayer), a=alat)
atoms.calc = EMT()
atoms.get_forces()
return atoms
# clean slabs
path_to_refs_db.unlink(missing_ok=True)
# connect to refs_db for writing
with connect(path_to_refs_db) as refs_db:
# connect bulk_db for reading
with connect(path_to_bulk_db) as bulk_db:
for row in bulk_db.select():
alat = row.cell[0, 1] * 2 # lattice constant
symb = row.symbols[0]
for nlayer in nlayers:
myid = refs_db.reserve(layers=nlayer, surf=symb, ads='clean')
if myid is not None:
atoms = clean_surface_reference(symb, alat, nlayer)
refs_db.write(
atoms, id=myid, layers=nlayer, surf=symb, ads='clean'
)
# isolated atoms:
for ads in ads_syms:
atoms = Atoms(ads)
atoms.calc = EMT()
atoms.get_potential_energy()
myid = refs_db.reserve(ads=ads)
if myid is not None:
refs_db.write(atoms, id=myid, ads=ads)
The previous code snippet saves those 24 reference energies (clean surfaces
and isolated adsorbates) in a refs.db file.
Suppose we want to extract the clean surfaces from
the refs.db and store them in clean_surfaces.db.
We could change the script and run the calculations again,
but we can also manipulate the files using the ase db tool. First, we
move over the clean surfaces:
$ ase db refs.db ads=clean --insert-into clean_surfaces.db
Added 0 key-value pairs (0 pairs updated)
Inserted 21 rows
If we want we can delete those from refs.db:
$ ase db refs.db ads=clean --delete --yes
Deleted 21 rows
Since this is only to demonstrate usage of the command line we will keep them.
Now we might want to extract the three atoms (pbc=FFF, no periodicity):
$ ase db refs.db pbc=FFF --insert-into isolated_atoms.db
Added 0 key-value pairs (0 pairs updated)
Inserted 3 rows
Finally, as we have not deleted anything these are the databases which we should have build so far:
$ ase db ads.db -n
63 rows
$ ase db refs.db -n
24 rows
$ ase db clean_surfaces.db -n
21 rows
$ ase db isolated_atoms.db -n
3 rows
Analysis#
Now we have what we need to calculate the adsorption energies and heights:
# connect to ads_db for updating
with connect(path_to_ads_db) as ads_db:
# connect to refs_db for reading
with connect(path_to_refs_db) as refs_db:
for row in ads_db.select():
# atoms
e_ads = refs_db.get(formula=row.ads).energy
# clean surface
e_clean = refs_db.get(
surf=row.surf, layers=row.layers, ads='clean'
).energy
ea = row.energy - e_ads - e_clean
h = row.positions[-1, 2] - row.positions[-2, 2]
ads_db.update(row.id, ea=ea, height=h)
Once run we can inspect the results for three layers of Pt:
$ ase db ads.db Pt,layers=3 -c formula,ea,height
formula| ea|height
Pt3C |-3.715| 1.504
Pt3N |-5.419| 1.534
Pt3O |-4.724| 1.706
Rows: 3
Keys: ads, ea, height, layers, surf
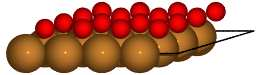
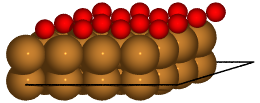
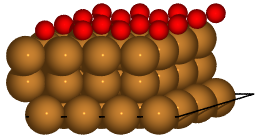
Finally, we can load specific structures of the database and create figures:
with connect(path_to_ads_db) as ads_db:
for nlayer in nlayers:
atoms = ads_db.get(surf='Cu', ads='O', layers=nlayer).toatoms()
atoms_sc = atoms * (4, 4, 1)
renderer = write(f'Cu{nlayer}O.pov', atoms_sc, rotation='-80x')
renderer.render()
Note
While the EMT description of Ni, Cu, Pd, Ag, Pt, Au and Al is alright, the parameters for C, N and O are not intended for real work!