
Note
Go to the end to download the full example code.
Fine tuning POV-Ray settings for high quality images#
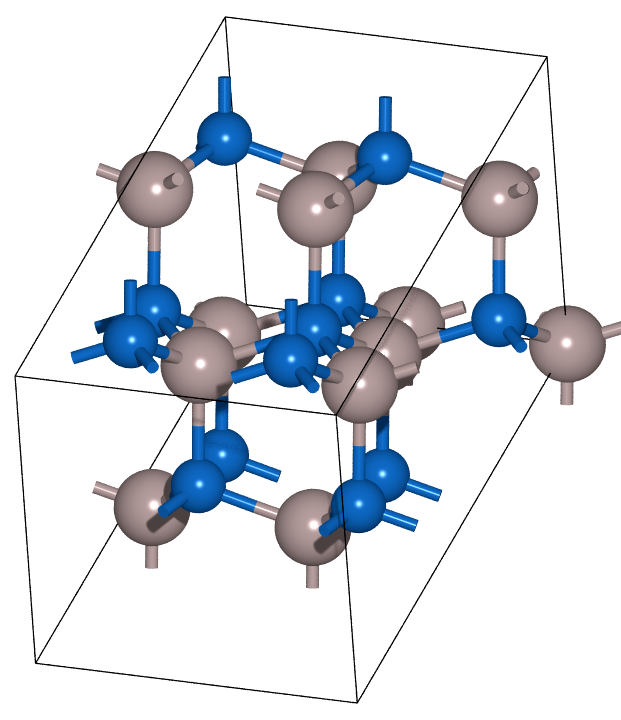
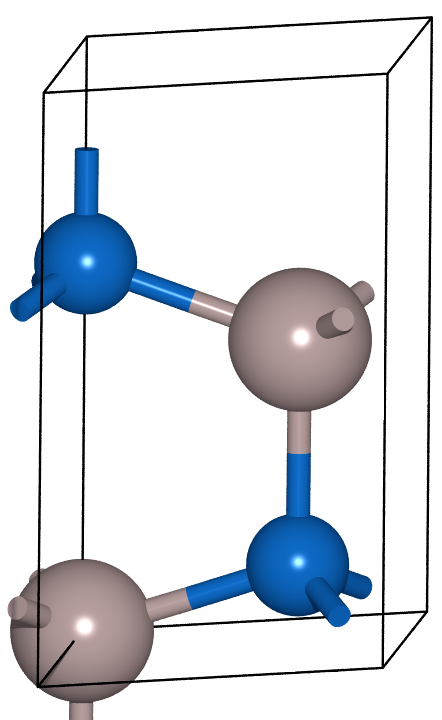
# This script creates pictures of sample structures, the cut sqs cell will keep
# the same crystallographic orientation in the image as the unit cell
from ase import build
from ase.data import covalent_radii
from ase.data.colors import jmol_colors as chemical_colors
from ase.io.pov import get_bondpairs, write_pov
unit_cell = build.bulk('AlN', 'wurtzite', a=3.129, c=5.017)
sqs_cell = build.cut(unit_cell, a=[1, 0, 1], b=[-2, -2, 0], c=[1, -1, -1])
names = ['unit_cell', 'sqs_cell']
list_of_atoms_obj = [unit_cell, sqs_cell]
# The rest are rendering settings
# see the gallery to see examples of the built-in styles
style = 'simple'
# reverts to jmol_colors if not unspecified
color_dict_rgb255 = {'N': [23, 111, 208], 'Ga': [230, 83, 17]}
# used to automatically guess bonds
covalent_radius_bond_cutoff_scale = 0.9
# for the rendering atom radii
radius_dict = {
'O': 0.8,
'Al': 0.6,
}
# for unspecified elements
radius_scale = 0.6
# use ASE GUI to find your perfect orientation
# rotation = '45x, -35.264y, 30z' # down <111> for a cubic structure
# rotation = '0x, 0y, 0z' # down z-axis
rotation = '37x, -79y, -128z'
# povray specific kwrds
kwargs = {
'transparent': True, # Transparent background
'canvas_width': None, # Width of canvas in pixels
'canvas_height': 720, # Height of canvas in pixels
# 'image_height' : 22,
# 'image_width' : 102, # I think these are in atomic units
# 'image_plane' : None, # Distance from front atom to image plane
# # (focal depth for perspective)
# 'camera_dist' : 170.0, # Distance from camera to front atom,
# 'camera_type': 'orthographic angle 35', # 'perspective angle 20',
# ultra_wide_angle
# 'area_light' : [(2., 3., 40.) ,# location
# 'White', # color
# .7, .7, 3, 3], # width, height, Nlamps_x, Nlamps_y
# 'point_lights' : [], # [[loc1, color1], [loc2, color2],...]
# 'background' : 'White', # color
'depth_cueing': False,
'celllinewidth': 0.01, # Radius of the cylinders representing the cell
}
# generic projection settings (passed to plotting variables)
generic_projection_settings = {
'rotation': rotation,
'show_unit_cell': 2,
# 'extra_offset':(2.0, 0.0)
}
# some nice helper functions
def make_radius_list(atoms, radius_dict, radius_scale=0.9):
per_atom_list = []
for z, symbol in zip(atoms.get_atomic_numbers(), atoms.symbols):
if symbol in radius_dict.keys():
per_atom_list.append(radius_dict[symbol])
else:
per_atom_list.append(radius_scale * covalent_radii[z])
return per_atom_list
def make_color_list(atoms, color_dict):
per_atom_list = []
for z, symbol in zip(atoms.get_atomic_numbers(), atoms.symbols):
if symbol in color_dict.keys():
per_atom_list.append(color_dict[symbol])
else:
per_atom_list.append(chemical_colors[z])
return per_atom_list
# converting RGB255 to RGB1 for povray.
color_dict = {}
for symbol in color_dict_rgb255:
color_dict[symbol] = [val / 255.0 for val in color_dict_rgb255[symbol]]
# loop over atoms objects to render them
for atoms, name in zip(list_of_atoms_obj, names):
radius_list = make_radius_list(
atoms, radius_dict, radius_scale=radius_scale
)
bondpairs = get_bondpairs(atoms, radius=covalent_radius_bond_cutoff_scale)
color_list = make_color_list(atoms, color_dict)
# These have to be set per-atom
kwargs['textures'] = len(atoms) * [style]
kwargs['colors'] = color_list
kwargs['bondatoms'] = bondpairs
# PlottingVariables needs the radii to set the image plane size
generic_projection_settings['radii'] = radius_list
pov_name = name + '.pov'
povobj = write_pov(
pov_name, atoms, **generic_projection_settings, povray_settings=kwargs
)
povobj.render()